As a bioinformatician, and especially as my time in Armenia was my elective for the scientist training program while I was a pre-registration clinical bioinformatician, the main focus of my volunteering was to carry out bioinformatics research. I did this at the Institute of Molecular Biology (IMB), where I joined the BIG Pathways project, of the BIG (bioinformatics group) group at IMB, which is headed by Professor Arsen Arakelyan, who is also the director of IMB.
A challenge in research is that getting into a project sufficiently to work on it effectively can take time, as there are new equipment, methods and collaborators to work with, as well as basic knowledge to acquire about the field to understand the work. As I only had 6 weeks I wanted to do something which I could quickly get into and make useful contributions, so when Arsen Arakelyan was explaining the projects that they were working on at the IBM I chose one that I could quickly be useful in that fit my knowledge and skills and provided immediately useful work for IMB.
For my project, I chose to get involved in a biological network mapping project, known as the BIG Pathways project. The nature of this project is that proteins and other biological molecules do not act in isolation, rather they form parts of interconnected systems that allow biological organisms to respond appropriately to different stimuli. Different components have different functions in these systems. Some proteins are cell surface membranes that interact with molecule outside the cell, others are transcription factors which start the transcription of genes responsible for response to particular stimuli, and between these two end points there are numerous signalling complexes along the way. The study of these networks is known as systems biology.
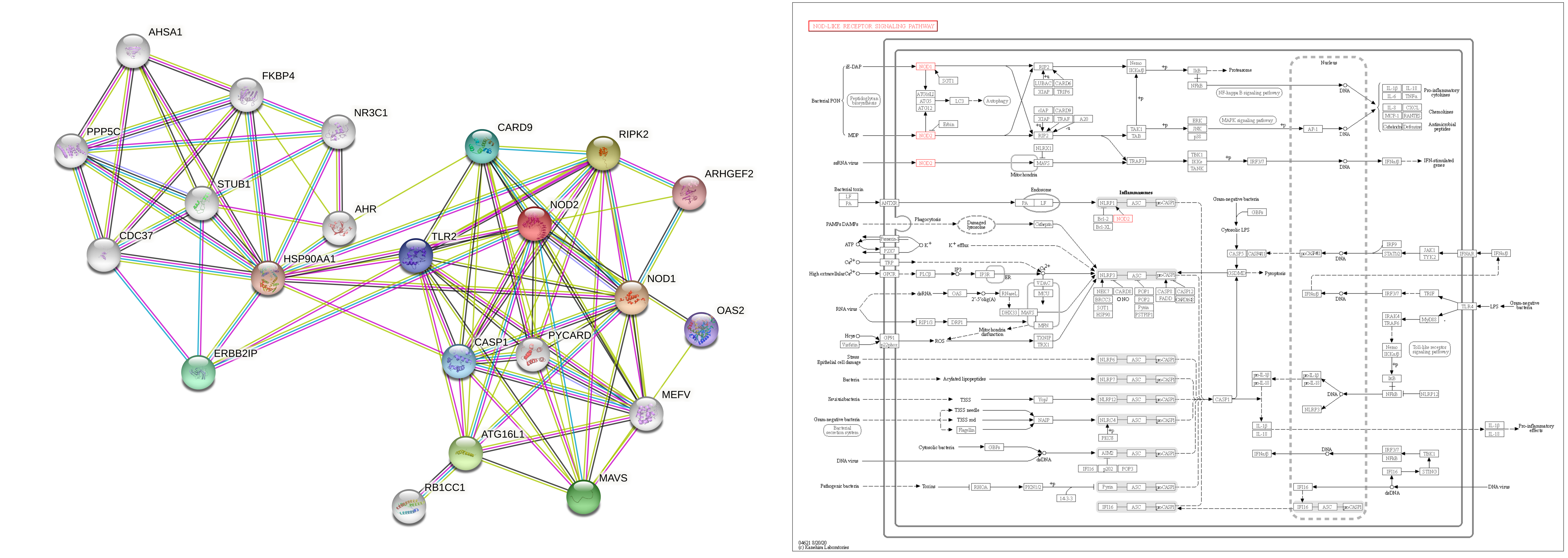
There are a lot of searchable databases describing the interactions between biological molecules (Figure 1). There are databases such as KEGG pathways which describe pathways, and mechanisms of action, and directionality in detail, and where each pathway can be searched from and retrieved as a whole. Another database is STRING, which is a large and flexible database which can be used to search by gene name, and the type of evidence used to determine that an interaction takes place. However a drawback of these extant databases is that they are often automatically produced by reference to other databases, or to literature. As a result, the evidence used to determine that an interaction takes place cannot be verified. Nor is the process by which fully constructed network maps are published able to be scrutinized.
What the BIG Pathways project at IMB was doing was producing a curated database of biological networks where evidence for interactions was assessed manually and logged into the database, which could be scrutinized in future. Interactions in the database were logged with the exact text and source describing the interaction and its evidence, thus the evidence for the interactions logged in the database would be verifiable by its users, and so could be appraised to determine if it was biologically relevant for the investigator’s research, and updated in light of new evidence that amends the literature record. Overall the idea behind BIG Pathways was to have a database that researchers in other groups at IMB could use in their own research, as systems biology is an essential tool for many modern post genomic research methods which rely on understanding what biological components do in biological systems.
I was given the NOD pathway to map, which was a pathway that is involved in the cellular response to infectious disease. The strategy I adopted was to use the KEGG pathways database to generate a rough outline of the pathway that I would build. KEGG had an outline of a pathway, which I would model the pathway I would build from. Once I had this outline of the pathway, I needed to locate evidence to support each reported interaction. To do this I used STRING, and with STRING users can select specific types of evidence to include when constructing networks, and view networks that have been constructed on the basis of the lines of evidence selected. To find the type of evidence I needed, I used “text mining”, which includes evidence derived from the same proteins being mentioned in the same publication. As I was looking for scientific literature describing the interactions, the text mining function was ideal for this requirement, however it has limitations, as I will describe.
This presented the first problem I had collating the evidence. The text mining search in the STRING database does not consider the context of their mention in the same text. Computers are generally unable to conceptualize the context of the proximity of two phrases, thus I a text mining search could return phrases like: "The ABCD1 is shown to interact with the EFGH2 gene through a protein fragment complementation assay" which would be useful for showing a biological connection. However the search could equally find A gene panel containing genes ABCD1, EFGH2, IJKL3 and MNOP4 was sequenced via Illumina Dye Sequencing" While the fact that both genes are included in a panel may suggest some sort of biological relationship between them (else why would a researcher be researching them), the fact they’re being tested together in a panel does not demonstrate any biological connection. It is important when appraising the text found to make sure text reporting interactions are of the first type as opposed to the second type.
Once the fact that an interaction was taking place had been determined by appraising the text, this presented a second challenge in appraising the evidence text. I had to consider what type of experiment was used to demonstrate the relationship between the molecules. An experiment that shows that two proteins interact in vitro does not necessarily mean the interaction occurs in vivo, it may be that the proteins are found in different parts of the cell, or are not expressed at the same time as one another, and therefore any interactions they have are merely theoretical. Furthermore some interactions between proteins are transient, and not long lasting, which makes them not especially amenable to experimental verification. There are though other experimental techniques, or more often combination of techniques which can determine if an interaction is taking place in vivo. The key to solving this puzzle was to assess what the literature was saying about a particular interaction and what evidence it was using to make that determination. It would be exhaustive to describe all literature that I read and explain what it meant, but a broad understanding of different experimental techniques and ability to read scientific literature was the key to addressing this challenge.
Another challenge for building the pathways was a philosophical one involving how to appropriately describe the interactions in the network. Many protein interactions involve many proteins forming a single complex, and the composition of this complex can vary, as does what each complex does. An example of this is the nodosome complex, which can be composed of multiple proteins, and each different composition has different downstream outputs from the complex. It then becomes a question of what is this complex called and how should it be logged in the database? Two ways of looking at this are: "These are all nodosomes, but nodosomes can be made of, and do different things" or "These are different types of nodosome which should be logged separately" The key to answering this philosophical challenge is consistency in how everyone is logging different protein complexes. Inconsistencies in how different types of interaction are logged are a problem with manually curated databases, as inconsistent data cannot be assimilated as easily, or analyzed programmatically. To ensure the consistency of logging interactions, the BIG Pathways project had a standard operating procedure describing how such ambiguities should be logged.
I found working on the BIG Pathways project was very interesting, and it also prepared me very well for one of my university courses about systems biology for clinical genetics which I took shortly after my time in Armenia. So it was a very useful hands on learning experience, as well as allowing me to effectively contribute towards one of the research at IMB. I found it and interesting paradox that although bioinformatics seeks to create workflows that can be used to analyze biological systems in a high throughput manner, the data quality can be limited by the need for manual curation of the input data, making the process of systems biology a trade off between the size of the data and the quality of the data. I found my strategy of refining data derived from large scale database searches to focus my searches for high quality data to be an effective one to combine these two competing factors.