Sequencing Methodologies
Sanger Sequencing
Sanger sequencing was the first type of sequencing methodology developed. In Sanger sequencing, the DNA sequence is replicated in a solution containing modified nucleotides known as di-deoxynucleotidetriphosphates (ddNTPs). These are different to normal nucleotides, once they have been incorporated into the DNA they do not allow further chain elongation, thus the DNA replication for the molecule they are incorporated into stops at that nucleotide.

This method was originally implemented with different ddNTPs corresponding to each of the nucleotides, and 4 reaction mixtures and only one form of ddNTP in each reaction mixture. These reaction mixtures would then be separated by agarose gel electrophoresis in 4 wells and the pattern of bands would indicate the DNA sequence. However the modern method attaches a different dye to each ddNTP and a single reaction mixture can be used. After the reaction, instead of being separated on agarose gel in multiple lanes, these can be separated by a single capillary gel. Due to the fluorescent tags, these can be detected by a laser detector. The rate at which the DNA fragments migrate through the capillary tube corresponds to their size, with smaller fragments migrating first. This means that the fastest migrating fragments are those which have had ddNTPs incorporated earlier, which means they are read from 5ꞌ to 3ꞌ, as this is the same order that the sequence elongates in. If the dyes attached to the ddNTPs are specific to the identity of the nucleotide that they replace, this means that each colour will correspond to a particular nucleotide, meaning that the series of colours indicates the series of nucleotides.
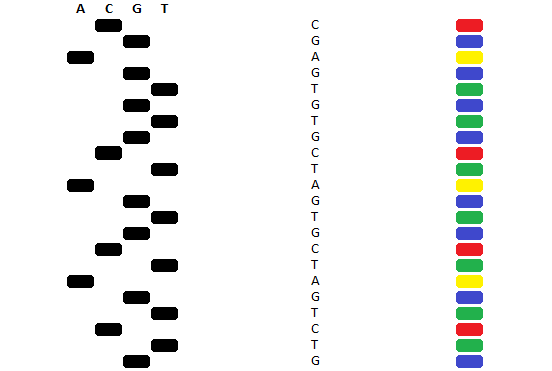
Modern Sanger sequencing technologies can read these sequences of colours and rapidly convert them into DNA sequences. The output of this process is a sequence chromatogram, which can be analysed by a number of pieces of software. Sanger sequencing is a simple and low cost protocol, however for many large sequencing applications it has been superseded by more modern Next Generation Sequencing technologies.
Next Generation Sequencing
Next Generation Sequencing describes sequencing technologies that are more modern than Sanger sequencing (in that they are the next generation of sequencing technologies). There are many such technologies available, but what they have in common is that they are more rapid than Sanger sequencing and are more amenable to high throughput sequencing. A next generation sequencing technology that is widely used is the Illumina Dye Sequencing method.
In Illumina Dye Sequencing, the DNA sample is randomly broken into fragments of different lengths using enzymes called transposomes. These fragments then have adaptors attached to them at either side of the fragment. These adaptors are crucial to the protocol, and so any fragments which do not have adaptors attached to them are discarded. After adaptors are added to the fragments, indices, primer binding sites and terminal sequences are attached to the fragments. Indices allow the sequences to be identified during data analysis. Primer binding sites are where the DNA replication that happens during sequencing starts from. Lastly terminal sequences allow the fragments to bind to the analysis cell.

Fragments are passed through a flow cell. This contains oligonucleotides (short pieces of DNA), which are attached to the flow cell. When the tagged DNA fragments enter the flow cell, they attach to these oligonucleotides via the terminal sequences of the tags. This binding takes place through complementary base pairing. Once the fragments are bound, a process of “bridge amplification” occurs. In this, DNA polymerase produces double stranded DNA from the fragment and the oligonucleotides in the flow cell, thus producing double stranded DNA with terminal sequences at either end. The original fragment sequence is discarded, and the other end of the single stranded DNA binds to another oligo forming a bridge. DNA polymerase then moves along this bridge, forming new double stranded DNA, with both ends attached to the flow cell. When the complementary pairings between these sequences dissociate, this leaves two strands from the original sequence fragment, doubling the amount of DNA that can produce signal in the later sequencing steps. As both of these strands can form other bridges with other unattached nucleotides, this step can be repeated over and over again to form clusters, which amplify the signal. As the cluster expansion process produces both forward and reverse strands, the reverse strands are removed before sequencing commences.
When the clusters are sequenced, they are not sequenced using an irreversible cap which terminates chain elongation, such as the ddNTP used in Sanger sequencing. Instead the sequencing uses a fluorescent tags which are attached to the nucleotides incorporated, a different colour for each nucleotide. During sequencing a cycle takes place of adding the nucleotides to the flow cell, which are then incorporated into the DNA fragments undergoing elongation during the sequencing, because these nucleotides are tagged, only one nucleotide can be incorporated in a cycle. Because the fluorescent tag on each nucleotide is distinct for each type of nucleotide incorporated, and a cluster will only be composed of identical sequences, this means that these clusters will fluoresce indicating that the fragment bound to them has incorporated a particular nucleotide in that elongation cycle.
The fluorescent tag can then be removed; this allows a new nucleotide to be incorporated into the sequence. Because the fluorescent tag removal, and the new nucleotide incorporation are separate processes, this means that the sequencing process is cyclical. This means that not only is the type of nucleotide incorporated into the fragment known, but so too is the location that it was incorporated. Consequently, the sequences of fluorescent tags detected at a particular cluster during sequencing cycles can be used to identify the sequence of the fragment at that cluster.
Once the clusters have been sequenced, these produce sequence contigs. These are short fragments of sequence that were produced during the random breaking of the fragments at the beginning of the sequencing process. This is in contrast to Sanger sequencing, where a single sequence covering the entire sample is produced. These contigs are overlapped using the analysis software to produce a sequence, or they can be aligned with a template sequence, which would be the case if the sequencing is being used to detect variants, as in gene panels.

Illumina dye sequencing is faster than Sanger sequencing, and also benefits from being massively parallel, being able to sequence a large number of samples in parallel, and analyse the sequencing contigs to sequence a large number of genes in parallel. This so called high throughput DNA sequencing has greatly reduced the cost of DNA sequencing and made DNA sequencing a growing tool for use in medicine. This means that the genetic basis for a patient's condition can be understood and from this personalised medicine can be developed for them.